 Published on Aug 12, 2025 by Arcadia Science
Published on Aug 12, 2025 by Arcadia Science Assembling and annotating an Asgard archaea and giant virus dataset of over 840,000 proteins
Contributors (A-Z)
Assembling and annotating an Asgard archaea and giant virus dataset of over 840,000 proteins
Purpose
We wanted to build a deeply annotated proteome resource to expand the phylogenetic breadth of our investigations into protein evolution and sequence–structure–function relationships. Therefore, we compiled and annotated the proteomes of Asgard archaea, the closest relative of eukaryotes, and giant viruses, which naturally infect many of the unicellular organisms we work with at Arcadia. This dataset should serve as a valuable community resource for scientists interested in protein evolution and the origin of eukaryotes.
We built this dataset from publicly available genome assemblies from NCBI, comprising 649 Asgard archaea and 446 giant virus entries. Three hundred eleven of the Asgard archaea and all 446 of the giant virus assemblies included proteomes, and we assembled and annotated the data from those. We chose not to annotate the assemblies without proteomes, so there's likely more to discover in public databases.
- Data from this pub is available on Zenodo.
- All associated code and critical data are available in this GitHub repository.
Share your thoughts!
Feel free to provide feedback by commenting in the box at the bottom of this page or by posting about this work on social media. Please make all feedback public so other readers can benefit from the discussion.
Background and goals
As a company, we want to explore the boundaries of protein sequence–structure–function relationships across the tree of life. So far, our explorations have spanned the breadth of eukaryotic diversity, enabled by the data underlying our organismal selection tool, “Zoogle” [1]. However, these boundaries could be further probed by expanding our analyses beyond the evolution of eukaryotes (Figure 1). Asgard archaea are the closest relatives of eukaryotes [2][3][4][5], making them a clear priority for extending our work deeper into evolutionary time. In addition to finding the edges of sequence–structure–function diversity, Asgard archaea encode much of the same cellular machinery as eukaryotes [5][6][7][8][9][10], and we hope this resource will enable a deeper understanding of the origins of eukaryotes. Giant viruses (or nucleocytoplasmic large dsDNA viruses, NCLDV) represent a similarly underdeveloped opportunity to study sequence–structure–function relationships. These viruses infect primarily single-cell eukaryotes, meaning their divergent proteins function in eukaryotic cells and perturb eukaryotic cell biology. Much of their proteome is "dark matter," with no sequence homology to anything in public databases [11][12][13].
Among these proteomes are many homologs of eukaryotic proteins associated with genetic disease — proteins involved in translation, DNA and RNA processing, metabolism, cytoskeletal architecture, and trafficking. Most research on Asgard archaea has focused on a limited number of these homologs of eukaryotic signature proteins like actins and ESCRTs [2]. To move further, we need an annotated dataset of largely uncharacterized proteomes to comprehensively characterize protein sequence, structure, and functional diversity across the tree of life.
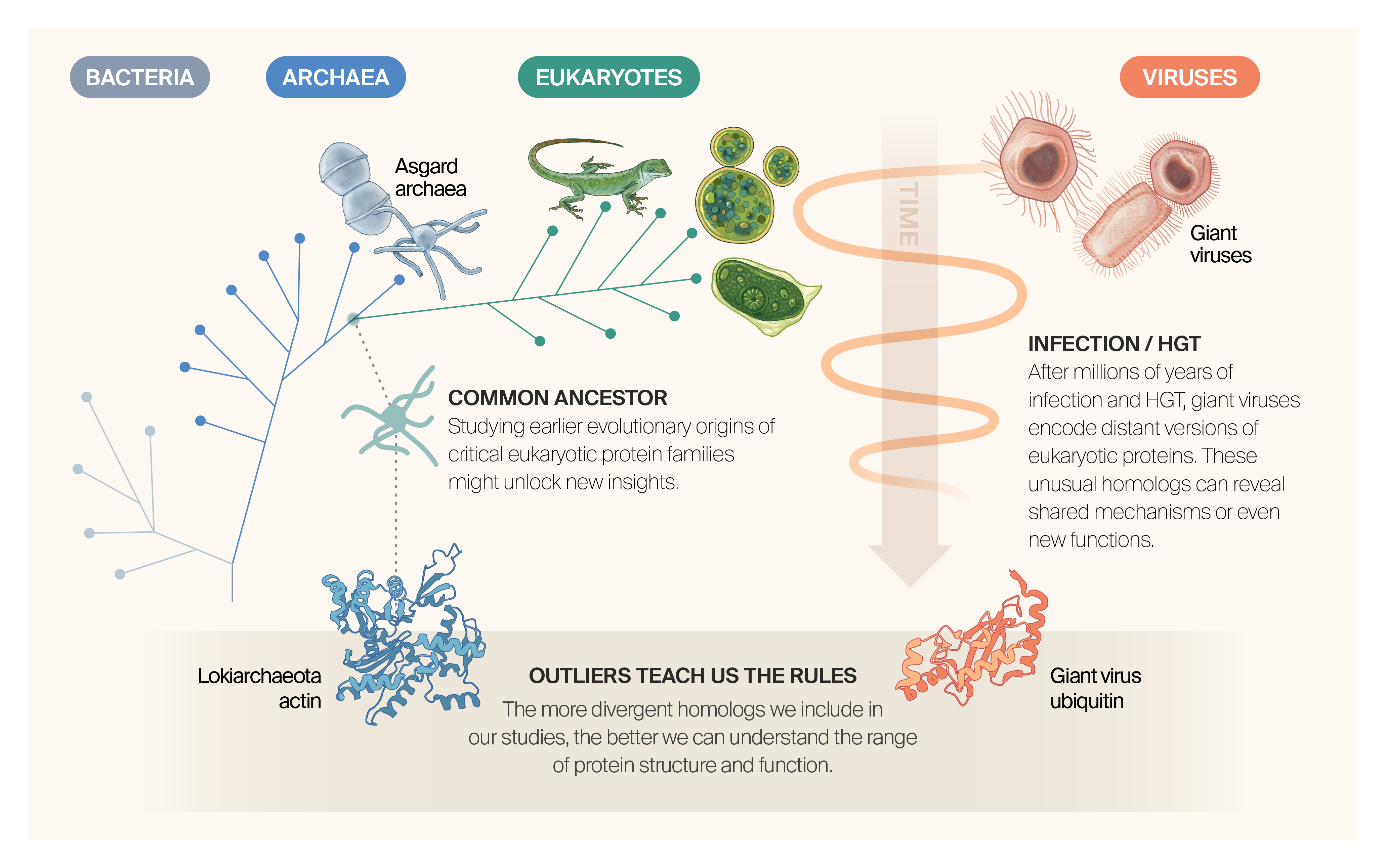
We should study non-eukaryotic genomes to get a more complete picture of sequence–structure–function relationships.
Eukaryotes originated from within Asgard archaea, suggesting that studying the Asgard proteome could offer novel insight into sequence–structure relationships deep into evolutionary time.
Giant viruses naturally infect single-cell organisms close to the root of the eukaryotic tree and have exchanged genetic information with their hosts continuously throughout time.
Specific goals
This work aligns with and facilitates an expansion of our research into protein evolution and design. We previously developed the ProteinCartography pipeline [14], and we used components of that tool to compile this dataset. We’ve shown previously that integrating sequence, structure, and functional data can unlock discoveries across large evolutionary time-scales, so we’re confident this is a practical approach to gain novel insights from the Asgard archaea and giant virus proteomes we’ve assembled here.
Given this context, our specific goals for this project were to:
- Create a comprehensive, consistently annotated database: Process 311 Asgard archaeal and 446 giant virus proteomes using the same pipeline, applying the same categorization rules and analysis parameters across > 840,000 diverse proteins.
- Characterize functional and structural landscapes: Map the distribution of proteins across functional categories and predict structural features like transmembrane domains, signal peptides, and intrinsic disorder. This characterization enables the prediction and prioritization of protein families and individual proteins for folding and functional annotation.
- Quantify evolutionary diversity within orthologous groups: Apply Hill’s diversity metrics and calculate average pairwise sequence identity (APSI) to identify patterns in how proteins evolve within orthologous families, revealing different evolutionary constraints across functional categories.
- Map connections to eukaryotic proteins: Use DIAMOND to perform homology searches against eukaryotic proteomes from the organisms included in our Zoogle organismal selection tool.
- Define the "structurally dark" proteome: Filter the dataset against structural databases such as PDB, AlphaFold DB, and ESMAtlas to identify proteins lacking structural characterization. This filtering provides understudied targets for future structural studies.
- Establish a foundation for targeted functional studies: Using domain architectures for each protein, evaluate their likelihood to produce high-quality predicted structures, setting the stage for future work.
Given our goal of exploring the boundaries of protein sequence–structure–function relationships across the tree of life, this dataset of > 840,000 Asgard archaea and giant virus proteins should serve as a crucial resource. By systematically annotating and analyzing these proteomes, we aim to deepen our understanding of how protein sequences dictate structure and how far sequences can diverge while maintaining fold and function. This knowledge will help us prioritize targets for structural and functional studies and enhance our work designing biologics and identifying disease targets.
The approach
For a visual overview of our approach, see Figure 2.
We downloaded proteomes from NCBI associated with 311 Asgard archaea and 446 giant virus genome assemblies. These assemblies span all known Asgard phyla (Prometheoarchaeota, Heimdallarchaeota, Thorarchaeota, Odinarchaeota, Lokiarchaeota, Hodarchaeota, Helarchaeota, Wukongarchaeota, Hermodarchaeota, and Njordarchaeota) and the major families of giant viruses (Mimiviridae, Phycodnaviridae, Ascoviridae, Marseilleviridae, Pandoraviridae, Pithoviridae, and assorted unclassified viruses). A substantial fraction of the Asgard assemblies belong to an “unknown” phylum, which we plan to probe in the future.
We filtered sequences to remove those with non-standard amino acids and ≥ 50% disorder, and standardized headers for consistent processing. Generally, we kept Asgard and giant virus proteins separated, running parallel analyses on each. We used OrthoFinder (v3.0; RRID: SCR_017118) [15] to identify and partition protein families, and Interproscan (v5.73-104; RRID: SCR_0058290) to characterize domain architectures [16]. We used USPNet [17] to identify signal peptides, predict subcellular localization, and define the mature protein sequences. We implemented a custom dictionary based on IPR codes and keyword matching to assign proteins to functional categories. We also screened each sequence against structural databases and conducted a Hill’s diversity analysis to characterize the diversity within orthogroups. We then conducted sequence-based homology searches against 63 eukaryotic proteomes corresponding to the organisms in our Zoogle organism selection tool. Finally, we integrated sequence features to calculate a normalized score (0–100) representing the likelihood that a protein sequence would produce a high-quality folding prediction. We implemented this pipeline using a variety of Python and bash scripts, as well as the Jupyter Notebook, “database_assembly.ipynb.”
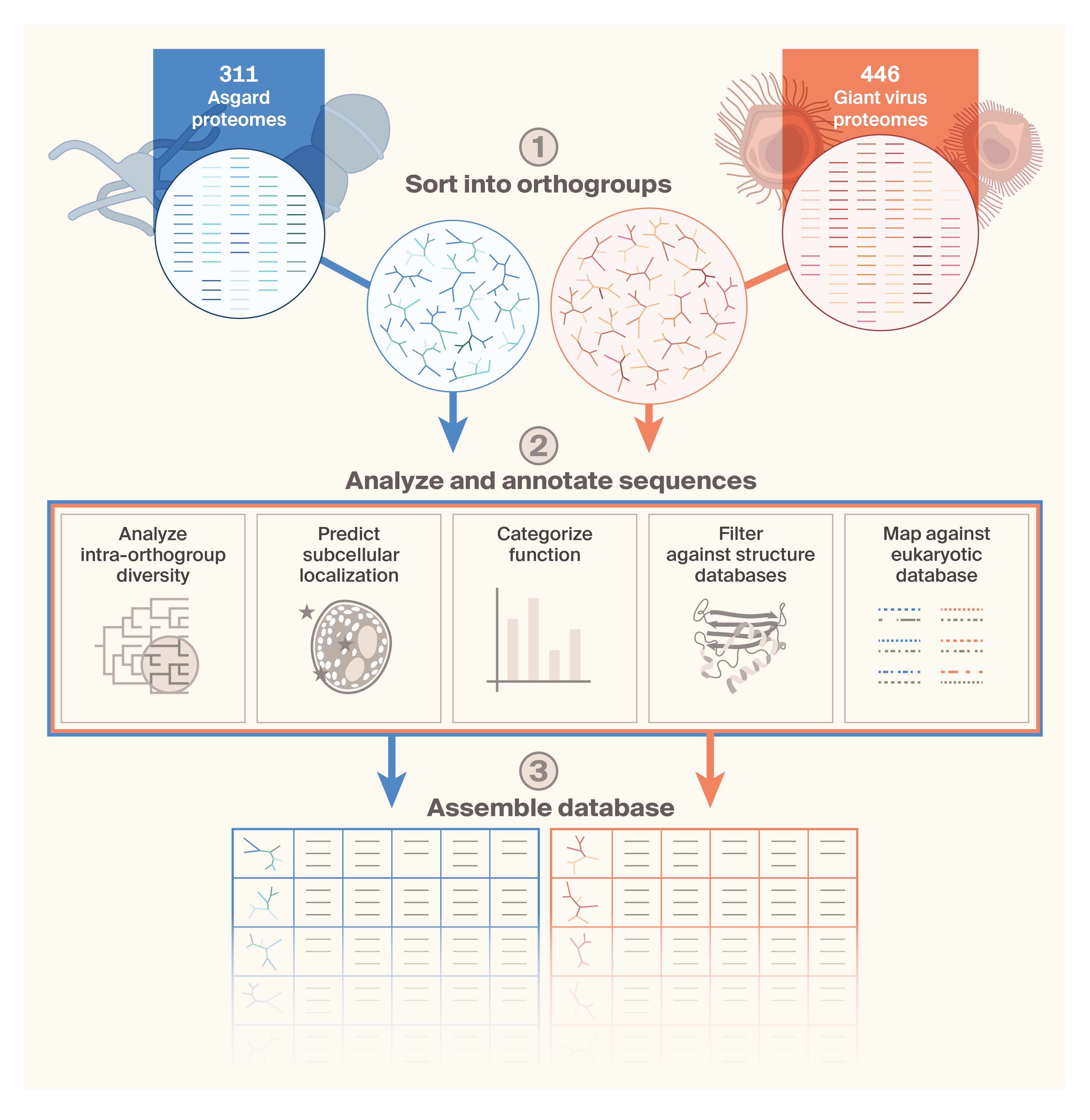
Schematic of the workflow we used to assemble the Asgard/giant virus proteome database.
We collected 311 Asgard and 446 giant virus proteomes and used OrthoFinder (v3.0) to sort them into orthogroups. We then comprehensively annotated the sequences using Hill’s diversity analysis, subcellular localization prediction, functional categorization, and filtering against structure databases. Finally, we queried the protein sequences against a custom DIAMOND database derived from 63 representative eukaryotic proteomes.
Orthology inference and diversity analysis
We used OrthoFinder (v3.0; RRID: SCR_017118) [15] to define orthogroups for the Asgard archaea and giant virus proteomes after filtering out sequences with non-standard amino acids and high disorder (> 0.5) using metapredict (v3) [18]. We then filtered to consider only orthogroups with more than five sequences in our diversity analyses, eliminating 10,611 orthogroups and left 11,613 encompassing 818,767 protein sequences out of the entire dataset of 844,750 proteins. We used MAFFT (v7.526; RRID: SCR_011811) [19] to align the sequences in each orthogroup and a highly parallelized version of FastTree 2 [20] called VeryFastTree (v4.0.5; RRID: SCR_023594) [21] to infer approximate maximum-likelihood phylogenies for each orthogroup. We then used a custom script (hill_diversity_analysis.py) to run a Hill’s diversity analysis and calculated the average pairwise sequence identity (APSI) for each orthogroup. A "high" (Hi) value for a given metric (APSI, Shannon entropy, or observed richness) indicated that the orthogroup’s value for that metric was in the top 25th percentile, whereas a "low" (Lo) value for a given metric showed that the orthogroup’s value was in the bottom 25th percentile. Combined Hi/Lo classifications are based on these percentiles for individual metrics.
Protein domain identification, localization, and functional prediction
To determine the putative function of protein sequences, we characterized domain architectures using Interproscan 5 (v5.73-104, RRID: SCR_005829) [16] in Docker. We used USPNet [17] to identify signal peptides, derive mature protein sequences, and predict subcellular localization. We used a custom dictionary to define and sort proteins into functional categories “Cytoskeleton,” “DNA Info Processing,” “RNA Info Processing,” “ESCRT/Endosomal Sorting,” “Membrane Trafficking/Vesicles,” “Ubiquitin System,” “N-glycosylation,” “Nuclear Transport/Pore,” “Translation,” “Signal Transduction,” and “Metabolism” in our “database_assembly.ipynb” Jupyter Notebook. We used metapredict (v3) to predict the intrinsic disorder of each protein sequence [18].
Structural database filtering
We conducted a series of searches against existing databases to determine whether structural information existed for any proteins in the dataset. We first retrieved UniProt IDs for sequences in the database and queried these against the PDB and AlphaFold databases. We then conducted sequence-based searches against these databases. Finally, we used MMseqs2 (v17.b804f) [22] to filter all the PDB/AFDB double-negative sequences against MGNify clusters, and filtered those hits against UniProt IDs reported recently to be present in ESMAtlas [23]. 225,704/844,750 proteins were present in one of these databases, with the vast majority (224,725) found in the AFDB. 619,873 sequences lack any structural information.
Eukaryotic homolog identification
We downloaded complete proteomes from NCBI corresponding to the 63 eukaryotes in our “Zoogle” organism selection portal. We concatenated these proteomes into a single FASTA and made a custom database using DIAMOND (v2.1.11; RRID: SCR_016071) [24]. We then queried the Asgard archaea and giant virus proteomes against this database with a minimum score of e ≤ 1e−10 to be considered a hit.
Intrinsic quality score calculation
Given that we intend this dataset to be a resource for exploring the boundaries of protein sequence–structure relationships, we wanted to determine how likely any given sequence was to produce a high-confidence structural model. To do so, we developed a customized, normalized (0–100) “intrinsic quality score” incorporating the following parameters:
# --- Intrinsic Quality Scoring Parameters ---
# Length (amino acids)
OPTIMAL_LENGTH_MIN = 80
OPTIMAL_LENGTH_MAX = 500
LENGTH_SCORE_OPTIMAL = 20
LENGTH_SCORE_SUBOPTIMAL_PENALTY = -10
# Disorder (percentage)
LOW_DISORDER_THRESHOLD = 20
HIGH_DISORDER_THRESHOLD = 50
DISORDER_SCORE_LOW = 15
DISORDER_SCORE_HIGH_PENALTY = -20
TMD_PENALTY = -30
NO_TMD_BONUS = 5
# Signal Peptide
HAS_SIGNAL_PEPTIDE_PENALTY = -5 # Small penalty for complexity
# Domain Architecture (Complexity)
# Number of domains
LOW_DOMAIN_COUNT_THRESHOLD = 3 # <= this number is good
HIGH_DOMAIN_COUNT_THRESHOLD = 6 # > this number is complex
DOMAIN_COUNT_LOW_BONUS = 10
DOMAIN_COUNT_HIGH_PENALTY = -10
# Bonus for single domain proteins
SINGLE_DOMAIN_BONUS = 5
Data integration and visualization
We integrated these analyses into a central database using pandas (v1.5.3; RRID: SCR_018214) in Python. We used Plotly (v6.0.1; RRID: SCR_013991) for comparative visualizations, as implemented in the “Figures_DB_Pub.ipynb” notebook.
Additional methods
We used Google Gemini 2.5 Pro (preview) for coding and describing methods. We used Claude 3.7 Sonnet (extended thinking) to help with early drafts. We also used Claude to review our code and selectively incorporated its feedback. We used Grammarly Premium to help copy-edit draft text to match Arcadia’s style and to clarify and streamline our writing.
Code, including database construction scripts, annotation pipelines, and analysis notebooks, is available in our GitHub repository (DOI: 10.5281/zenodo.16597599). Access our raw data files on Zenodo (DOI: 10.5281/zenodo.16809414).
Findings about the dataset
Proteins with no structural information dominate our dataset; many proteins don’t have identifiable domains
Our database is derived from 311 Asgard archaea and 446 giant virus proteomes and contains 844,750 proteins in total — 736,919 from Asgard archaea and 107,830 from giant viruses (Figure 3, A–B). The Asgard archaeal proteomes are dominated by Heimdallarchaeota (~23% of proteins), Prometheoarchaeota (~29%), and Thorarchaeota (~17%), with a large fraction classified as unknown phylum (23%). The giant virus proteins primarily derive from viruses in the Mimiviridae (46%) and Phycodnaviridae (19%) families, while ~14% of proteins were from unclassified viruses.
Next, we analyzed the dataset to determine how many proteins we could assign putative or even hypothesized functions based on the sequence alone. Approximately 70% of Asgard archaeal proteins and 99% of giant virus proteins lack structural information based on filtering against the PDB, AlphaFold database, or ESMAtlas. 47% of Asgard proteins and 75% of giant virus proteins contained no protein domains identifiable by InterProScan. 67% of Asgard proteins and 82% of viral proteins didn’t return any eukaryotic hits from DIAMOND searches. Finally, 26% of Asgard proteins and 67% of viral proteins were “triple negative” across all three categories (265,084 proteins), so we’ll have to fold these to understand what they do and how we might use them in our work.
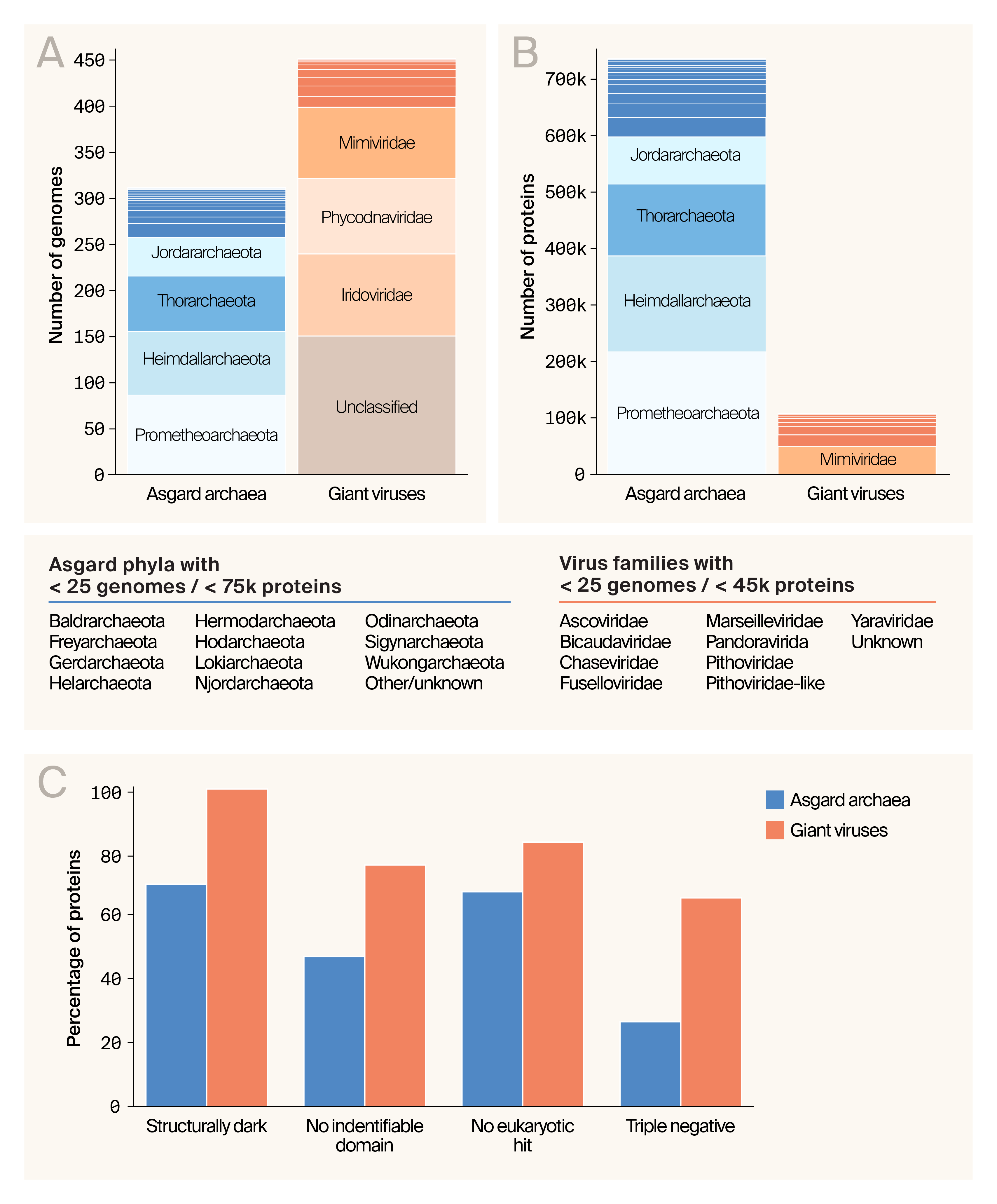
The database contains over 840,000 Asgard and giant virus proteins, many of which lack structural or functional annotation.
(A) The number of Asgard and giant virus genome assemblies represented in the dataset is stratified by Asgard phyla and virus families.
(B) Number of individual protein sequences comprising the dataset, again stratified by Asgard phyla and virus families.
(C) The percentage of proteins in Asgard and giant viruses that lack structural information, identifiable protein domains, and eukaryotic homologs, or all three.
Most proteins are cytoplasmic and involved in core cell biological functions
We predicted the subcellular localization of Asgard and viral proteins based on identifiable signal peptides, and both groups were remarkably similar. 97% of proteins in the database have no known signal peptide and are predicted to be cytoplasmic. Just under 3% are secreted, and we’d expect a small fraction, 0.2%, to be membrane-bound (Figure 4, A). Given that the intrinsic folding score we calculated included penalties for signal peptides and transmembrane domains, this breakdown suggests we can generate high-confidence structural models across the database.
The functional landscape (Figure 4, B) across Asgard and viral proteins is similar, though with some differences. Both groups contain a large percentage of metabolic, signal transduction, and DNA-processing proteins, but the Asgard proteome is particularly enriched in metabolic proteins. A much smaller percentage have only “general protein features,” meaning InterProScan identified domains, but they were too nonspecific to assign to a category.
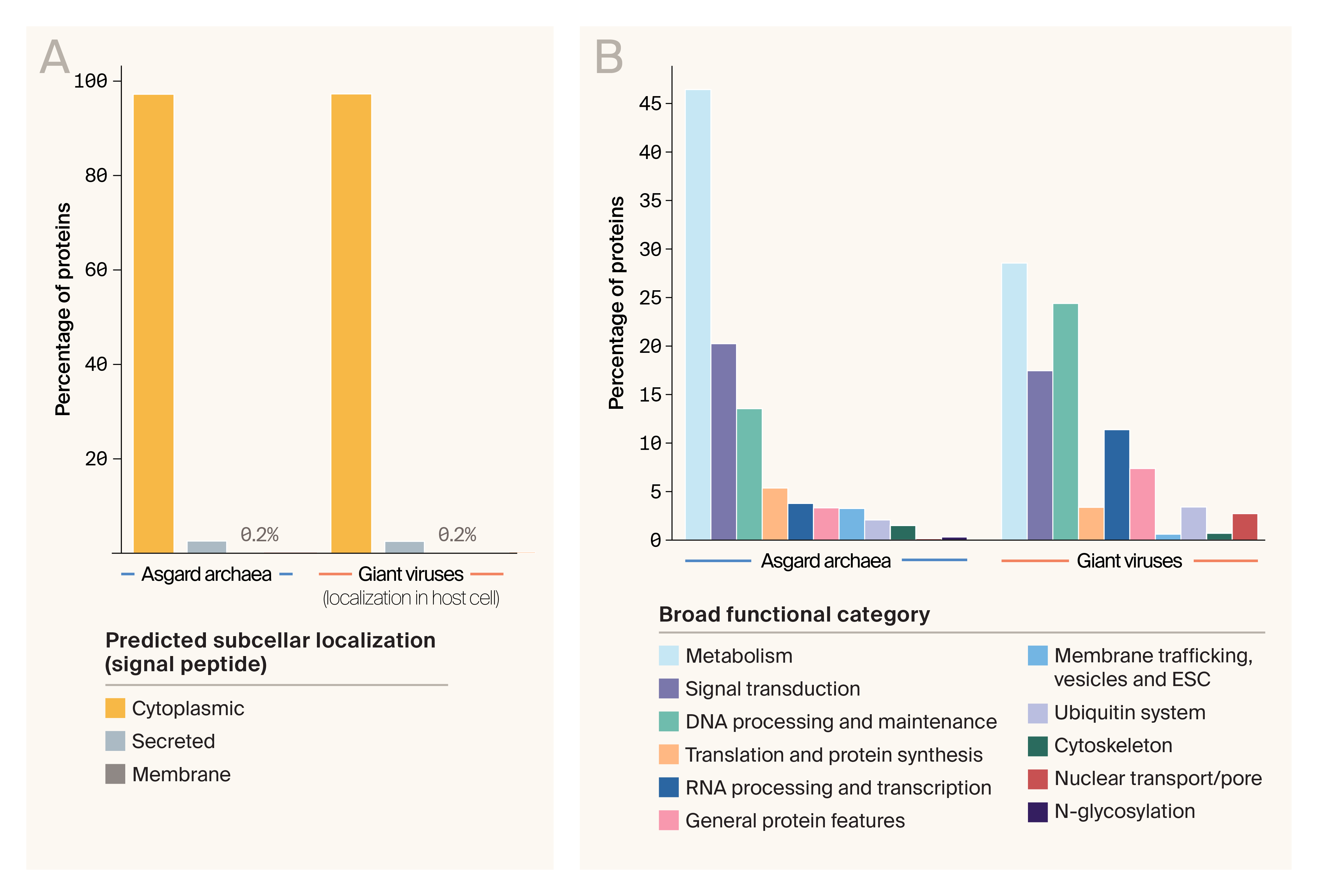
Predicted subcellular localization and functional categorization of the Asgard/giant virus dataset.
(A) Subcellular localization predictions for Asgard archaea and giant virus proteins.
(B) Functional categorization for Asgard archaea and giant virus proteins, based on IPR codes
Sequence conservation differs with predicted function, but phylogenetic breadth and sequence divergence correlate
To understand how proteins evolve within families, we analyzed Hill’s diversity [25] to characterize the evolutionary diversity within the 11,613 orthogroups containing ≥ 5 sequences. Specifically, we measured two key aspects of diversity: Shannon entropy, which captures how broadly distributed proteins are across the evolutionary tree (with higher values representing greater phylogenetic diversity in the orthogroup), and average pairwise sequence identity (APSI), a measure of how much the amino acid sequences in the orthogroup have diverged over time.
We expect these metrics to be inversely related, such that phylogenetically diverse orthogroups should exhibit lower APSI than orthogroups with only a narrow evolutionary range of organisms represented. Orthogroups that break this expected pattern would be particularly interesting. If Shannon entropy is high and APSI is also high, that would suggest the protein family is under purifying selection, with its function susceptible to changes in sequence. In contrast, a lower-than-expected APSI might suggest a protein family where the structure and function are relatively insensitive to the amino acid sequence conservation.
Shannon entropy and APSI were negatively correlated (Figure 5, A). We stratified orthogroups by whether they fall into each metric's tails (bottom 25th or top 25th percentile) and identified those in two tails (Figure 5, B), since these orthogroups are most interesting to us. Finally, we examined whether different functional categories are enriched in high-interest categories (Figure 5, C). Most functional categories are enriched for Hi entropy/Hi APSI, consistent with purifying selection on these protein families involved in core cellular functions. Strikingly, metabolic protein families show the opposite pattern; they're enriched for low entropy/low APSI, suggesting they have more sequence space available to explore without losing their essential functions.
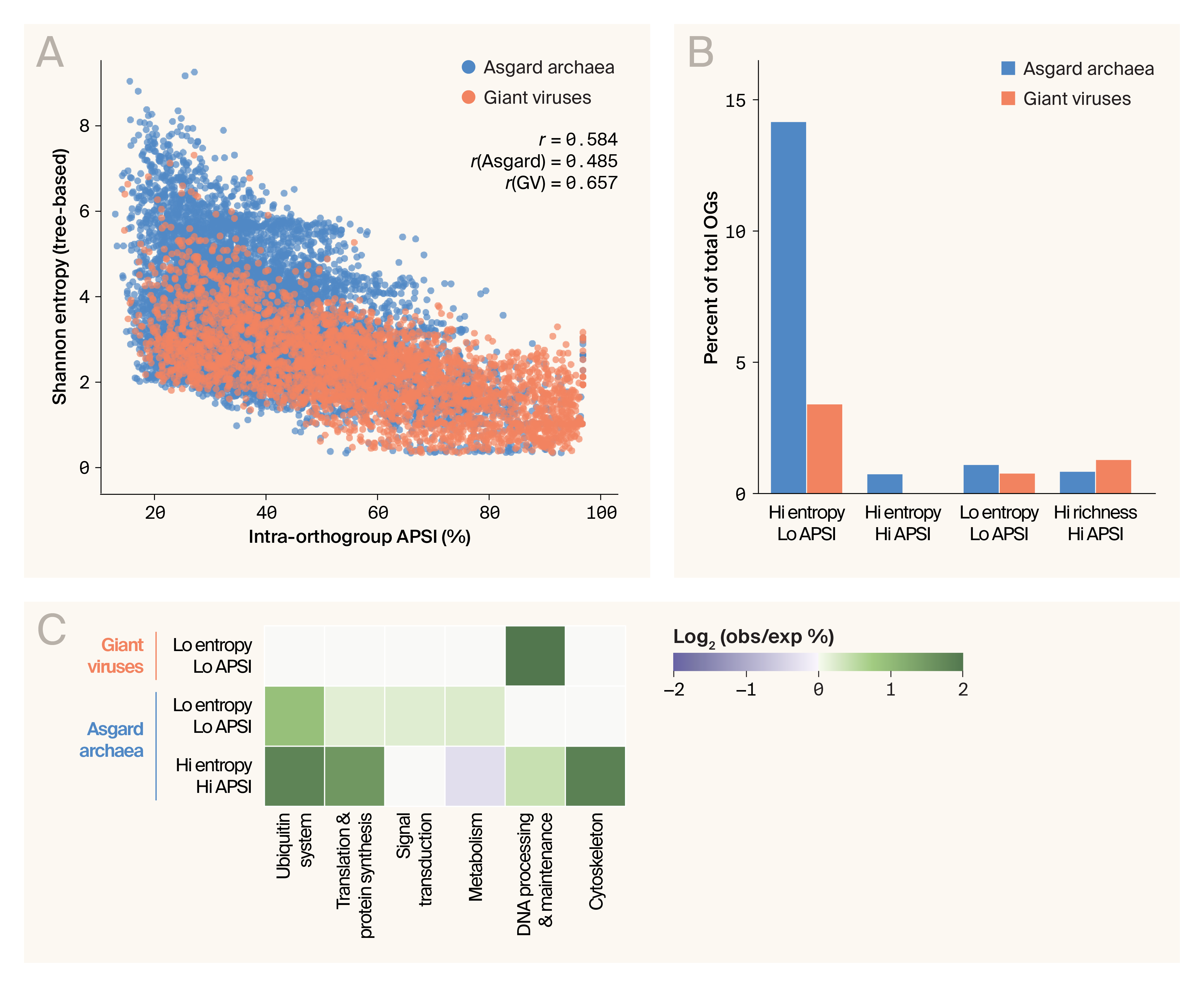
Orthogroups in the dataset exhibit varied sequence and structural diversity.
(A) Shannon entropy of each orthogroup plotted against the average pairwise sequence identity, showing a moderate negative correlation.
(B) Fraction of orthogroups that fall into the tails of Shannon entropy and APSI distributions.
(C) Heatmap showing the entropy/APSI profile of orthogroups in core functional categories. obs = observed; exp = expected.
Unicellular eukaryotes dominate among homologs
We were particularly interested in the potential to functionally annotate experimentally interesting proteins in unicellular eukaryotes we study in the lab, such as Chlamydomonas reinhardtii, Chlorella vulgaris, and others we identified via our Zoogle organism selection tool as potential model organisms for monogenic disease [26]. To facilitate this and assist with preliminary functional annotation, we assembled a custom DIAMOND database from the complete proteomes of the 63 organisms in Zoogle, which span 1.5 billion years of eukaryotic evolution. Then we determined which organisms appeared most frequently as top hits. For Asgard proteins (Figure 6, A), single-celled eukaryotes such as amoeba and green algae dominated the top hits, which met our expectations given these organisms and Asgard archaea are near the root of eukaryotic phylogeny. We’re particularly excited to see one of our most-used model organisms, Chlamydomonas reinhardtii, in the top five. We think it’s a promising platform for the functional annotation of many proteins. Other tractable organisms (e.g., Tetrahymena thermophila and Candida albicans) are also highly ranked, so we think there’s potential to take these proteins into the lab too.
The viral proteins similarly hit most frequently to unicellular organisms (Figure 6, B), which makes sense since those are the organisms they infect. We’re excited again to see C. reinhardtii in the top ten, but the presence of ciliates T. thermophila and Paramecium tetraurelia is particularly intriguing. So far, no ciliate viruses have been described in the literature, though some metagenomic datasets hint at the possibility [27][28]. Our results suggest extensive horizontal gene transfer between giant viruses and ciliates, so infection of those organisms by viruses related to those in our dataset seems likely.
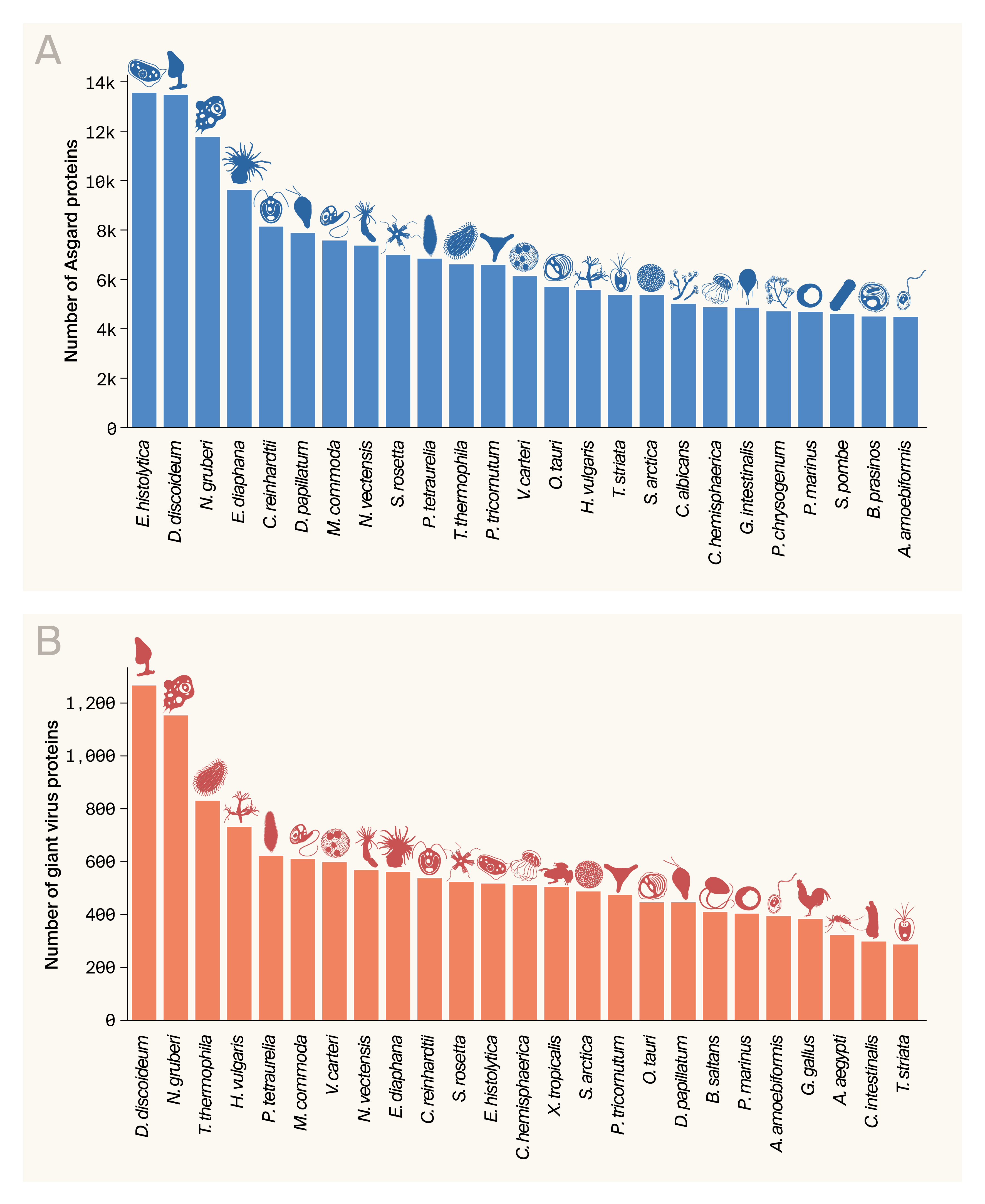
Top Zoogle organisms appearing as DIAMOND hits.
Zoogle eukaryotes appearing as top hits against Asgard proteins (A) or giant virus proteins (B).
You can access our proteome dataset, annotation files, and diversity metrics on Zenodo (DOI: 10.5281/zenodo.16809414). We’ve included the central database file and key supporting analyses for researchers exploring these proteomes.
Key takeaways
We hope this dataset, comprising more than 840,000 proteins from Asgard archaea and giant virus proteomes, will be a substantial new resource for probing the frontiers of protein sequence–structure–function relationships and evolutionary biology.
The database contains:
- Functional and structural characterization of 311 Asgard and 446 giant virus proteomes
- Evolutionary relationships within protein families
- Connections to eukaryotic proteins
- Metrics on the likelihood that a protein will fold computationally with high confidence
70% of Asgard and 99% of giant virus proteins lack structural information in public databases such as PDB, AlphaFold DB, or ESMAtlas. This structural darkness is compounded by the fact that a substantial fraction — 47% of Asgard and 75% of giant virus proteins — contain no identifiable protein domains via InterProScan, and 26% of Asgard and 67% of viral proteins qualify as "triple negative," meaning they have no structural data, no identifiable domains, and no detectable eukaryotic homologs. This vast unknown space highlights an untapped reservoir of information about protein sequence, structure, and function. There may even be novel protein folds or unexpected horizontal relationships within this dataset, which we’ve just begun to scratch the surface of.
Among the proteins in the dataset we can annotate, there’s functional enrichment for proteins involved in core cellular processes, including metabolism, signal transduction, and DNA/RNA processing. Asgard proteomes show a particular enrichment in metabolic proteins.
Our intra-orthogroup Hill’s diversity analyses revealed a largely expected negative correlation between Shannon entropy and average pairwise sequence identity, but pulling out high-interest groups revealed some interesting patterns. Specifically, most of the major functional groups are probably under purifying selection, with higher-than-expected sequence conservation given the phylogenetic diversity present in the dataset. However, metabolic protein sequences appear to be under a more relaxed constraint. Given the importance of metabolic pathways to disease, we’re excited to extract novel protein features central to core cellular functions from this data.
While large portions of both proteomes lack homologs in our Zoogle-derived DIAMOND database, those connections that do exist are to unicellular eukaryotes. Asgard proteins show strong links to protists like amoebae and green algae (including the model organism Chlamydomonas reinhardtii), reflecting archaea’s phylogenetic position near the root of eukaryotes. Similarly, viral protein homologs suggest unicellular eukaryotic hosts, and the prevalence of ciliates — previously not known to host archaeal viruses — among top homologs is a tantalizing hint of undiscovered viral diversity.
Next steps
This extensively annotated dataset opens numerous avenues for research to expand our understanding of protein evolution and structure–function relationships. We’ve identified several high-priority directions for our future work:
- We'll systematically explore the boundaries of sequence–structure–function relationships within the dataset. We’ll identify orthogroups with members with structural and, where possible, functional information in the literature and explore their diversity using a diverse evolutionary toolkit. This will allow us to start defining generalizable rules for how far and in what ways different protein families can diverge while retaining their necessary functions.
- We'll move beyond computational prediction to experimental validation of selected targets. The strong connections we identified to model organisms like Chlamydomonas reinhardtii and Tetrahymena thermophila provide excellent heterologous expression and functional characterization opportunities. We'll prioritize proteins that show unusual evolutionary patterns (such as high conservation despite high phylogenetic diversity) and those with potential connections to human disease-relevant pathways.
- We plan to conduct deeper evolutionary analyses to better understand the connections between Asgard archaea and eukaryotes. We’ll use phylogenetics to study specific protein families' evolutionary histories and trajectories, particularly those implicated in eukaryogenesis. We’ll also look for novel eukaryotic homologs in our structural predictions. These analyses will help illuminate how these proteins evolved and diversified across divergent lineages, potentially revealing new insights into the origins of eukaryotic cellular complexity.
- Finally, we'll continually refine and expand this dataset as new genomes become available. The 338 Asgard archaeal assemblies we identified but didn't process (due to a lack of proteome files) represent an immediate opportunity to expand our coverage. Additionally, integration with other Arcadia datasets will enable cross-domain comparative analyses that could reveal broader patterns in protein evolution and innovation.
Beyond our research, we hope that structural biologists, evolutionary geneticists, protein engineers, and microbiologists will find this dataset valuable for their investigations. We're eager to hear from researchers using it to explore protein structure prediction in highly divergent sequences, investigate the origins of eukaryotic cellular complexity, or discover novel enzymatic functions for biotechnology applications. We particularly hope this resource will accelerate research into the "structurally dark" proteome, where novel folds and functions likely await discovery. We welcome feedback from the community about other compelling research directions this dataset might enable.
We look forward to seeing how it contributes to our collective understanding of protein evolution across the deepest branches of the tree of life.
References
Share your thoughts!
Feel free to provide feedback by commenting in the box at the bottom of this page or by posting about this work on social media. Please make all feedback public so other readers can benefit from the discussion.
