Microscopy images are incredibly information-dense, and as a scientific community we are just beginning to streamline ways to more fully take advantage of this rich data. Our team’s ultimate goal is to figure out how to best capture, quantify, and compare more information across a broader range of species. In other words, how do we go from a picture being worth a thousand words to a million?
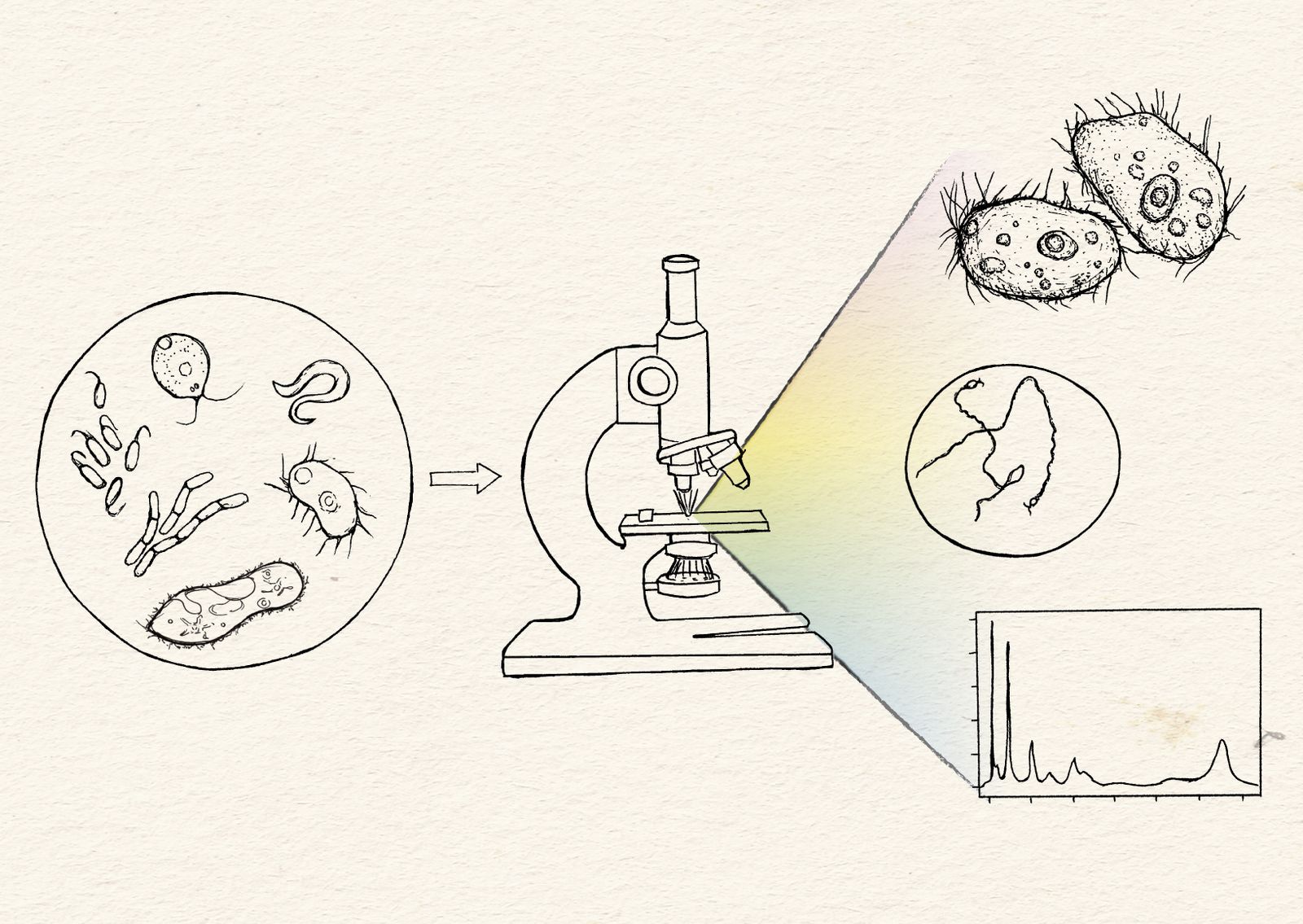
To carry out our vision for microscopy, we need to maximize exploration across the tree of life, enable informative comparisons, and expand our impact beyond Arcadia alone. As we build out our imaging center, we’re trying to set up systems that unlock the areas of biology that we’re currently pursuing. Namely, we’re optimizing our first systems for live and fixed imaging of primarily microorganisms. Methods-wise, we’re focusing on label-free imaging and using fluorescent dyes to track specific subcellular structures in organisms with limited or no genetic tools. We’ll update our strategies and focus areas on this page as we go.
As we chart out our future efforts, we’ve defined some core values that guide our decisions:
- Maximize exploration across the tree of life.
Why: Most of the organisms we study at Arcadia lack genetic tools. We’re actively developing methodology that will work well for a large swath of organisms rather than a select few. We may miss some opportunities because of this, but our overarching goal is breadth.
How: Our initial efforts aim to maximize label-free methods that don’t require genetic tools but still provide high-dimensional data. To take advantage of these, we need to develop microscopy-based proxies that can quickly clue us into species that may have the underlying biological traits we’re interested in. We’ll also improve sample preparation
- Increase capacity to capture diverse, complex phenotypes.
Why: Nature provides us with a library of phenotypes and functional solutions. To fully leverage these, we need to understand the design principles that drive these differences. To understand differences, we must be able to meaningfully compare biological properties across diverse species and conditions. These comparisons will generate hypotheses that we can test in the lab.
How: We’re developing screening platforms that maximize our ability to increase throughput and allow for the greatest reproducibility when comparing multiple conditions within and between species. We want to generate innovative and easy solutions for creating high throughput imaging assays to unlock new biology. As an example, we’re exploring multiple methods for organism confinement without the need of microfluidics or laser trapping [1]. In parallel, we’re developing computational methods that systematically parse these dense data in systematic ways beyond our manual abilities
- Optimize for impact beyond Arcadia.
Why: Arcadia alone will never explore at the scale required to fully maximize the utility and wonder of biology. We want to make sure our work catalyzes efforts outside of Arcadia too. While there is always a need to push resolution and speed, we can make the largest and most unique immediate impact by developing scalable, cost-efficient methods that others can borrow and build upon in the broader ecosystem.
How: To ensure our work can scale beyond Arcadia, we’ll focus on tools that give us the most bang for our buck. Also, our workflows and outcomes will be open and accessible to the rest of the scientific community. We’re currently innovating around high-throughput sample preparation and implementation of high-dimensional downstream data analyses. We’ll use commercially available microscopes rather than building custom setups.
Here at Arcadia, microscopy is a central platform that provides us with a lens to a mostly hidden world. Our team works closely with project scientists to help make their projects better versions of themselves while we simultaneously improve our own abilities in experimental design, biological imaging, and image analysis. Our first efforts have centered around getting maximal use out of commercially available microscopes that are best suited for live imaging so that our solutions can be recapitulated by others.
We currently have the following imaging systems in our facility:
-
Nikon Ti2-E & Yokogawa CSU W1-SoRa
-
Dual Orca Hamamatsu BT Fusion cameras, equipped with a LIDA light engine for color imaging with transmitted light
Biology happens in real time and across scales. Spinning disk confocal microscopes are an instrument of choice for optimizing signal-to-noise in live samples with less photobleaching compared to traditional laser scanning confocals. The SoRa mode increases spatial resolution (XY resolution to 120 nm). Here are some examples we’ve captured using the SoRa mode and a membrane dye (FM 4-64FX) to visualize cell membranes (left, middle) and cilia (right) in the ciliate, Colpoda steinii.
Super-resolution imaging of Colpoda steinii cysts.
Scale bar, middle = 10 µm, right = 500 nm.
-
Nikon Ni-E scope
-
Kinetix sCMOS camera
-
DS10 camera for color imaging
Our second microscope is optimized for high-throughput phenotyping of cell motility and capturing subcellular dynamics at high speed, such as flagellar beating dynamics or cilia in motion, through the use of a Photometrics Kinetix camera. Here is an example of using differential interference contrast (DIC) imaging at high speed to visualize the ciliary beating of the ciliate, Paramecium multimicronucleatum.
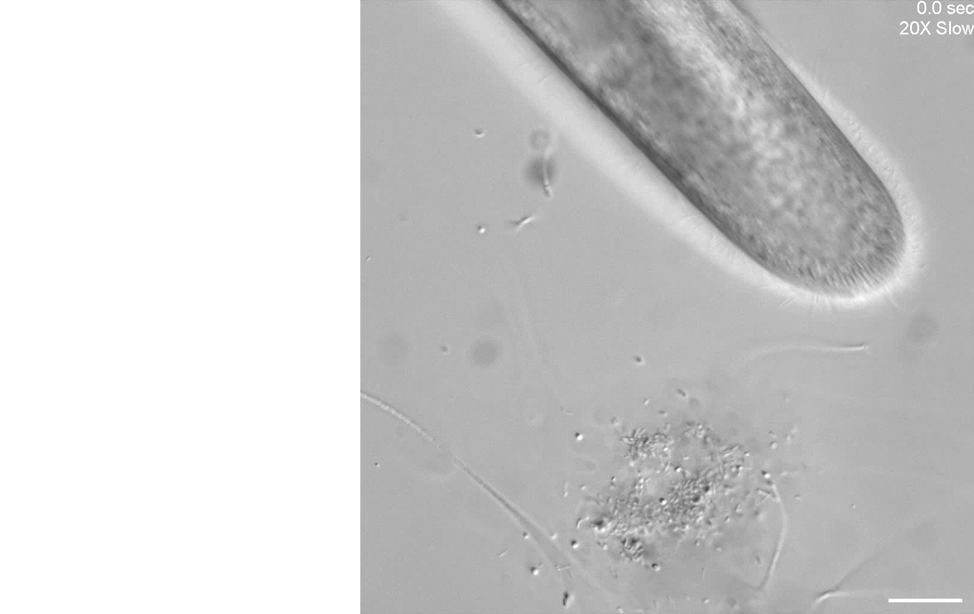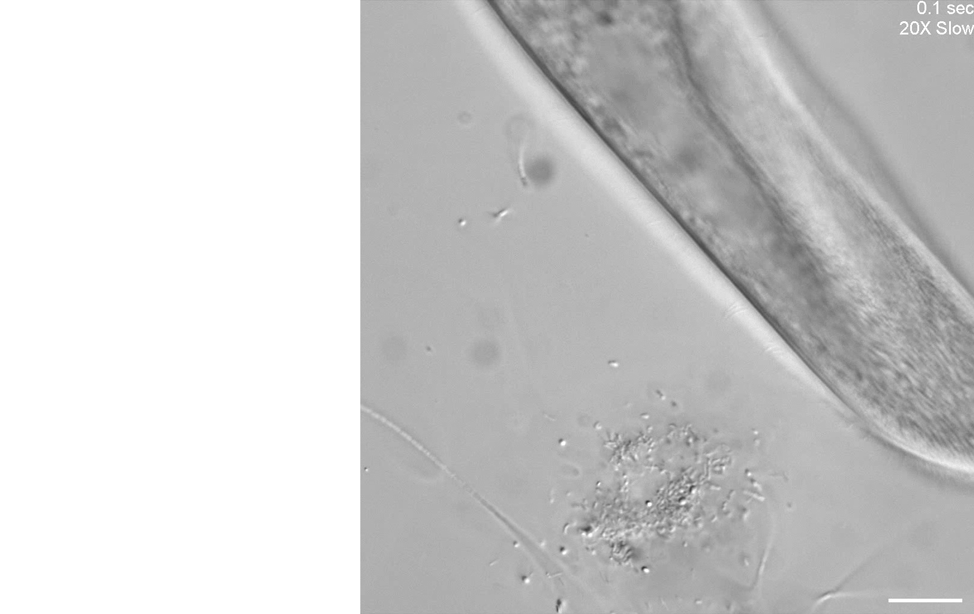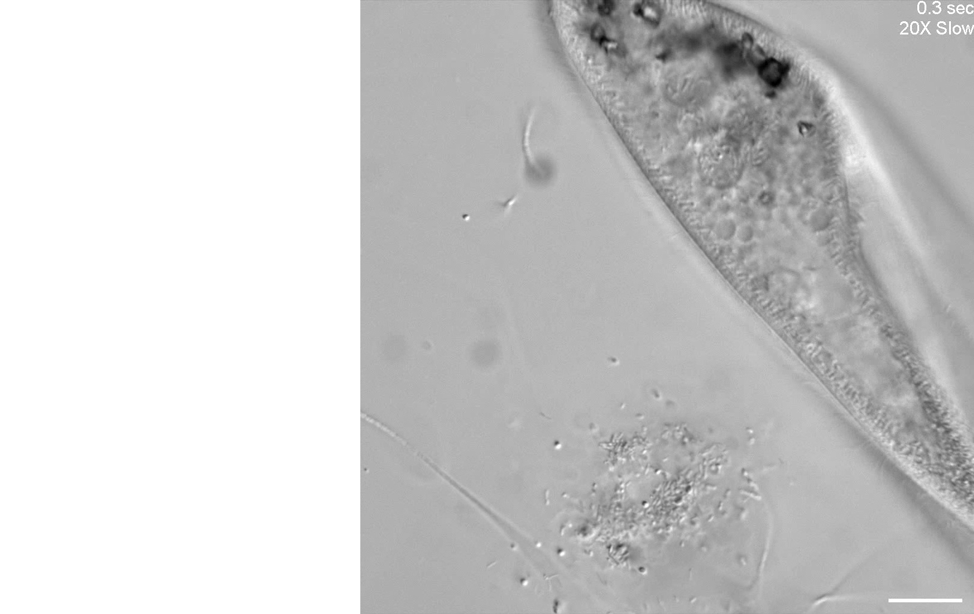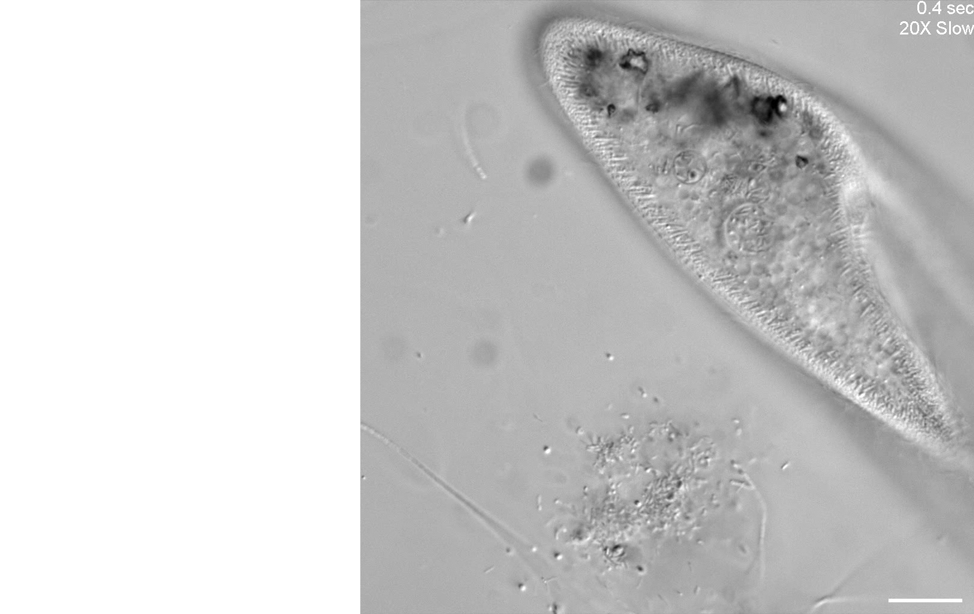
Unknown microorganisms and Paramecium multimicronucleatum.
Scale bar = 50 µm.
-
Leica STELLARIS 8
-
White light laser (WLL)
-
Coherent Raman scattering (CRS; includes both stimulated Raman scattering and coherent anti-Stokes Raman scattering)
-
FALCON FLIM (FAst Lifetime CONtrast system for Fluorescence Lifetime Imaging Microscopy)
As every good microscopist will tell you, sample preparation is critical for getting the most out of any imaging system. We decided to start by creating simple tools for live imaging of motile organisms between 5 and 300 microns in length.
We wanted to observe swimming cells with complex life cycles under different conditions but immediately struggled to image the cells for the hours-long periods needed because they are motile and readily swim out of focus.
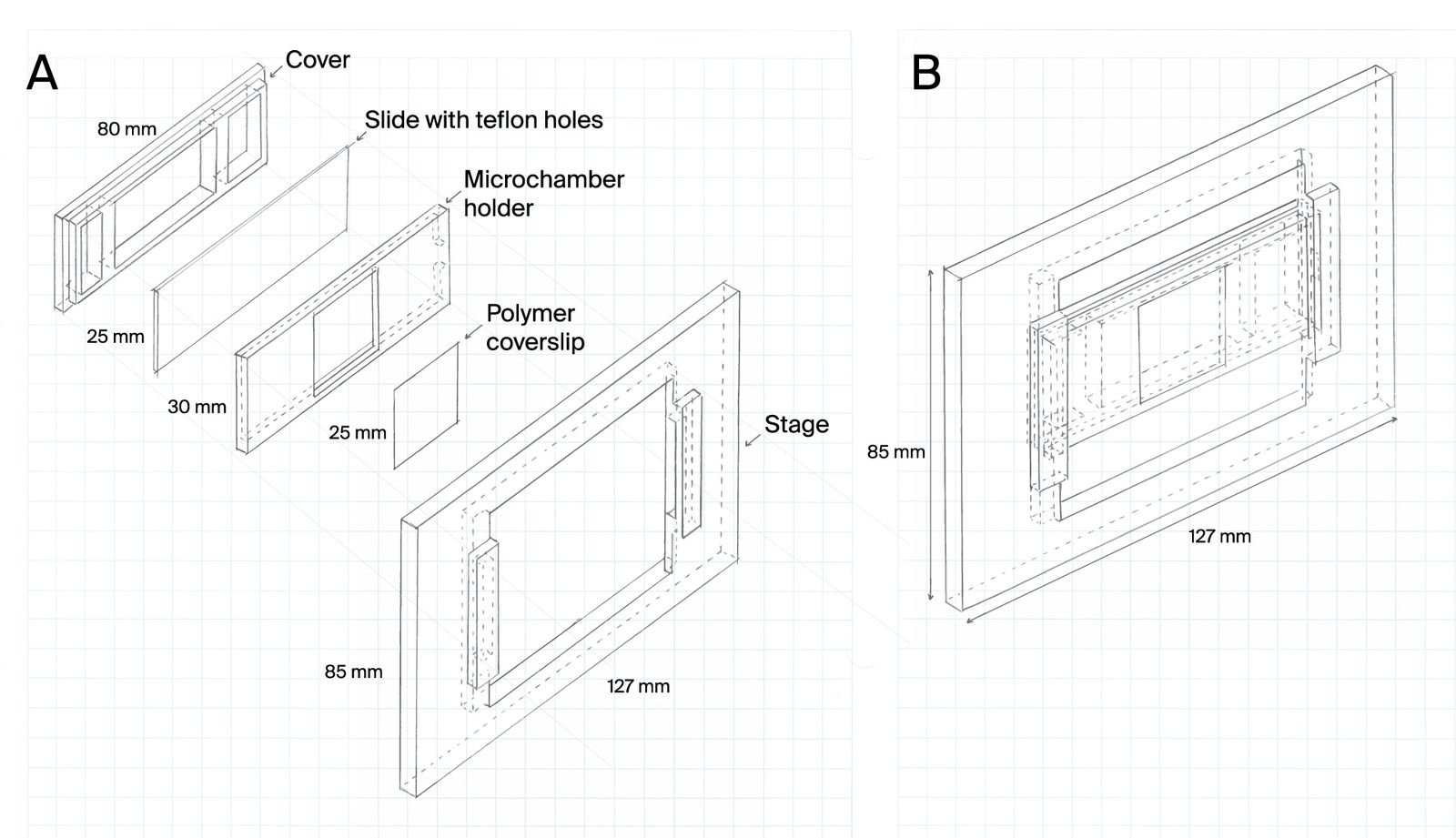
To address this, we first developed microchambers that confine cells within the field of view. The microchambers enhance the probability that a cell will be in the field of view at any given time and ensure that cells are maintained in locations visible under the microscope.
Learn more about this resource and try it yourself:
Microchamber slide design for cell confinement during imaging
Cells can be highly motile, moving in and out of a microscope’s field of view. Understanding complex life cycles is difficult without continuous observation. To overcome this challenge, we’ve developed a 3D-printed microchamber device to confine cells for long-term visualization.
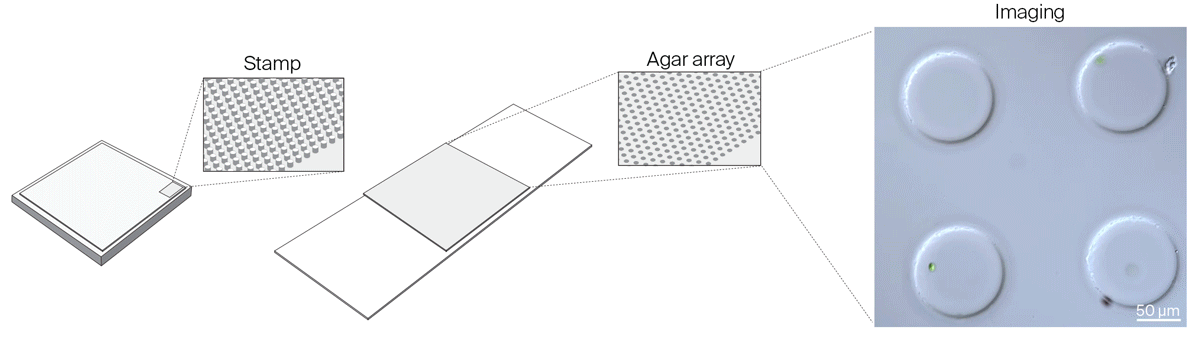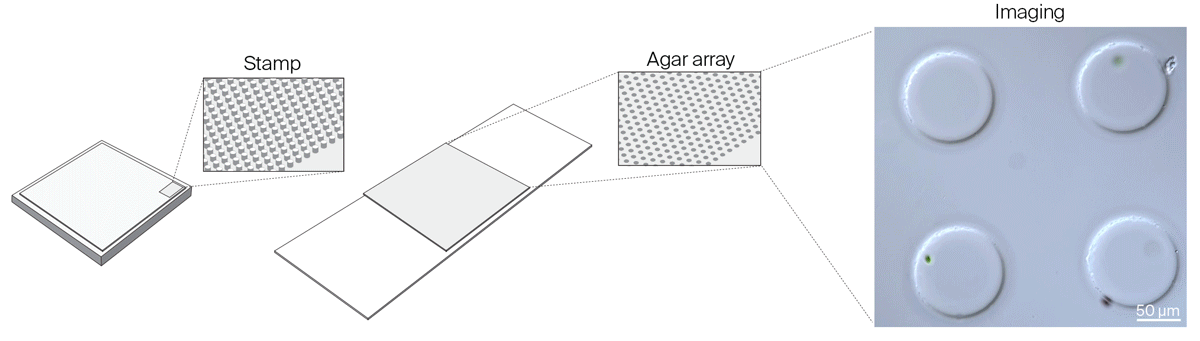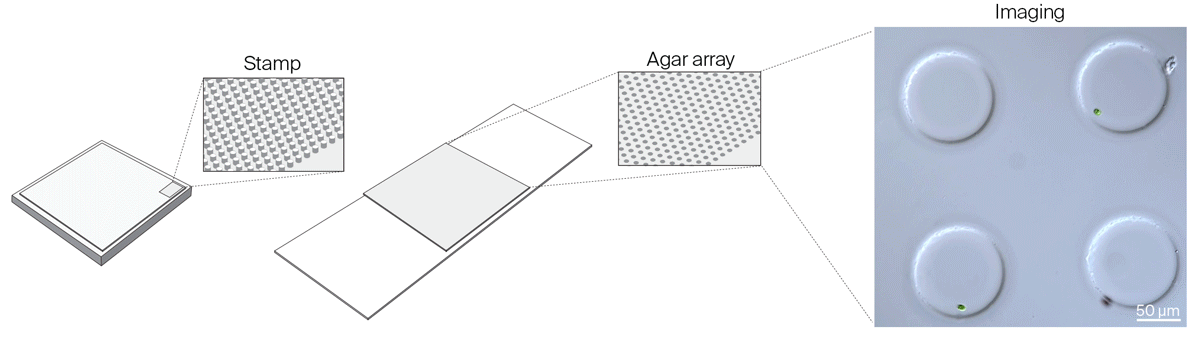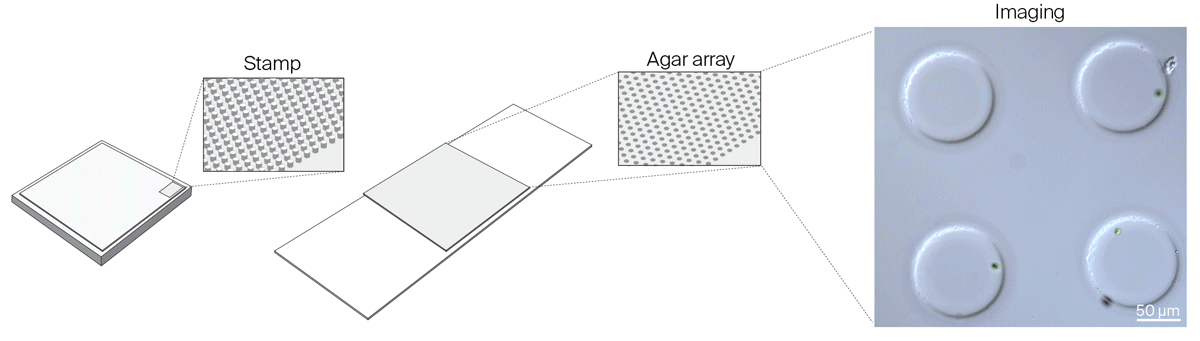
We realized that we needed greater flexibility in microchamber design to accommodate organisms of different sizes, to reliably capture isolated single cells in microchamber wells, and to improve cell tracking in all three dimensions. We settled on a new method to make microchambers using agar and pre-made PDMS stamps to restrict the movement of microorganisms in the x, y, and z dimensions. Read about these agar microchambers and how to make them yourself:
Gotta catch ‘em all: Agar microchambers for high-throughput single-cell live imaging
Constraining motile microorganisms for live imaging often requires costly microfluidics or optical traps to keep them in view. We used patterned stamps and agar to make versatile, inexpensive “microchambers” and offer a way to predict the right chamber size for a given organism.
Microscopy datasets can be large and unwieldy. To tackle this problem, we enriched datasets for smaller, more useful subsets. We implemented and compared three different feature-detection metrics that we can use to enrich data for in-focus frames — variances of the raw image, the Sobel-filtered image, and the Laplacian-filtered image. Our findings revealed that Sobel- and Laplacian-based methods significantly outperformed the raw pixel intensity variance approach in accuracy (as determined by manual expert annotations). Laplacian filtering tended to work better for DIC data, while Sobel filtering might be useful for cell and shadow tracking. For more details and to apply these methods in your research, check out our pub:
Streamlining microscopy datasets by enriching for in-focus frames
We distilled label-free microscopy data by comparing and implementing feature-detection algorithms. Sobel and Laplacian methods outperformed pixel intensity variance in accuracy.
Live imaging of swimming cells can provide high-dimensional data, but images can be difficult to acquire and analyze in high throughput. To address this problem, we developed a data acquisition and analysis workflow (SwimTracker) that increases the throughput and versatility of our previous sample preparation approach [1]. We show that this approach enables robust quantitative readouts of motility even without isolating single cells.
A high-throughput imaging approach to track and quantify single-cell swimming
Live imaging of swimming cells can yield insight into an organism’s viability and responses to environmental stimuli. We developed a microscopy workflow and image analysis pipeline, SwimTracker, to track motility phenotypes from swimming cells in high throughput.
Expanding our motility analysis to multicellular organisms let us capture biological traits that emerge from the coordinated behavior of several cell types in tissues or organs, which unicellular models can’t capture. Toward this goal, we developed an end-to-end experimental and computational workflow based on the motility-tracking software Tierpsy Tracker
A high-throughput imaging approach to track and quantify single-cell swimming
We used straightforward microscopy and computational analyses to reproducibly characterize a nematode motility phenotype with interpretable features. This method should be scalable for high-throughput phenotypic screening.
We’re interested in exploring label-free imaging at Arcadia, as many biologically intriguing organisms lack genetic tools. We’re currently looking at commercially available systems for doing fluorescence lifetime imaging microscopy (FLIM) and Raman spectroscopy.
We recently purchased a Leica Stellaris 8 confocal and coherent Raman scattering (CRS) system to explore label-free imaging with some of our in-house organisms. Here, we’re showing some optical sections and a 3D reconstruction of the trophic (motile) form of C. steinii, taking advantage of the organism’s autofluorescence to generate a high-resolution image (left). We also captured FLIM data using Leica’s TauSense feature for C. steinii trophic cells (middle) and cysts (right), showing that we can spectrally separate the cell wall based on autofluorescence intensity and fluorescence lifetime information (right). We’re pursuing questions around the high-dimensional signal from Raman and FLIM modalities and their utility for biological experimentation.
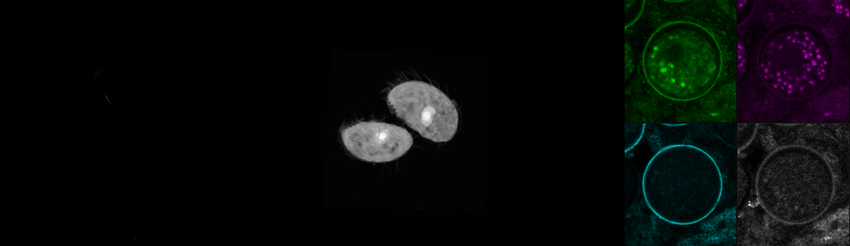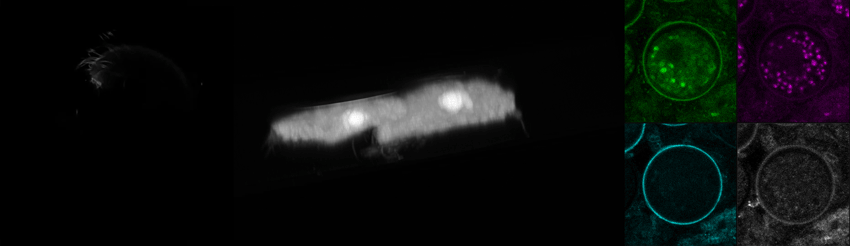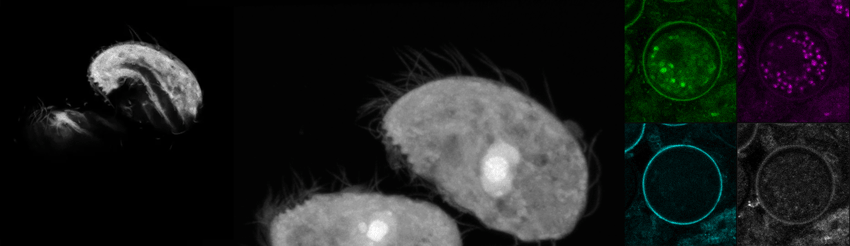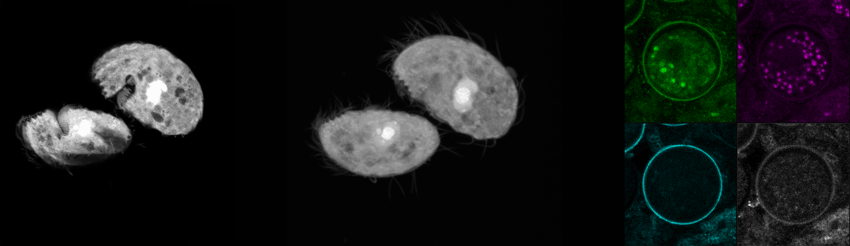
Autofluorescence and fluorescence lifetime imaging of C. steinii reveals sub-cellular structures.